 |
|

Theory research in the lab
Theory research involves formulating and solving mathematical motivated by consideration of biological scenarios, and interpreting the mathematical results for their contributions to biology. Advances in our theoretical work often focus on mathematical models, involving construction and analysis of new models, derivation of new results about existing models, development of new techniques for analyzing models, and model comparisons. Progress can also come from mathematical analyses of statistical methods, numerical studies and simulations, or introduction of new theoretical principles. We are also interested in the ways that theory research motivated by biological problems can advance mathematics itself.Theoretical research in the lab includes the following topics, among others:
- Mathematical properties of
population-genetic statistics
- Theoretical population genetics of
admixture
- Human migration and spatial expansion
- Consanguinity, identity by descent, relatedness, and runs of homozygosity
- Combinatorics of
evolutionary trees
- Coalescent theory
- Inference of species trees under gene tree discordance
Mathematical properties of population-genetic statistics. Many of the statistics used in population genetics are functions of the allele frequencies at a locus, a discrete set of nonnegative numbers that sum to one. This feature of allele frequencies contributes to surprising phenomena affecting some of the most popular population-genetic statistics, such as homozygosity and heterozygosity, the FST measure of genetic differentiation, and the r2 statistic for linkage disequilibrium. For example, the upper and lower bounds on homozygosity vary as a function of the frequency of the most frequent allele at a locus, the upper bound on FST varies with the homozygosity of a locus, and both upper bounds depend on the number of distinct alleles at a locus — all in a way that can be viewed as an epiphenomenon of the mathematical properties of the statistics. To facilitate sensible biological interpretations of observations of these statistics, we have been exploring their mathematical properties. This mathematical work provides explanations for a number of peculiar patterns seen in past applications of the statistics to population-genetic data.
- BK Moon, NA Rosenberg (2026) Integer sequences for diversity statistics. Journal of Integer Sequences 29: 26.1.5. [Abstract] [PDF]
- X Liu, Z Ahsan, NA Rosenberg (2025) Using mathematical constraints to explain narrow ranges for allele-sharing dissimilarities. Theoretical Population Biology 166: 116-137. [Abstract] [PDF]
- ML Morrison, KS Xue, NA Rosenberg (2025) Quantifying compositional variability in microbial communities with FAVA. Proceedings of the National Academy of Sciences USA 122: e2413211122. [Abstract] [PDF] [Supplement]
- TD Gress, NA Rosenberg (2024) Mathematical constraints on a family of biodiversity measures via connections with Rényi entropy. BioSystems 237: 105153. [Abstract]
- X Liu, Z Ahsan, TK Martheswaran, NA Rosenberg (2023) When is the allele-sharing dissimilarity between two populations exceeded by the allele-sharing dissimilarity of a population with itself? Statistical Applications in Genetics and Molecular Biology 22: 2023004. [Abstract] [PDF]
- ML Morrison, NA Rosenberg (2023) Mathematical bounds on Shannon entropy given the abundance of the ith most abundant taxon. Journal of Mathematical Biology 87: 76. [Abstract] [PDF] [Supplement]
- ML Morrison, N Alcala, NA Rosenberg (2022) FSTruct: an FST-based tool for measuring ancestry variation in inference of population structure. Molecular Ecology Resources 22: 2614-2626. [Abstract] [PDF] [Supplement]
- N Alcala, NA Rosenberg (2022) Mathematical constraints on FST: multiallelic markers in arbitrarily many populations. Philosophical Transactions of the Royal Society B: Biological Sciences 377: 20200414. [Abstract] [PDF] [Supplement]
- SM Boca, L Huang, NA Rosenberg (2020) On the heterozygosity of an admixed population. Journal of Mathematical Biology 81: 1217-1250. [Abstract]
- IM Arbisser, NA Rosenberg (2020) FST and the triangle inequality for biallelic markers. Theoretical Population Biology 133: 117-129. [Abstract]
- NA Rosenberg, DM Zulman (2020) Measures of care fragmentation: mathematical insights from population genetics. Health Services Research 55: 318-327. [Abstract]
- JTL Kang, NA Rosenberg (2019) Mathematical properties of linkage disequilibrium statistics defined by normalization of the coefficient D=pAB-pApB. Human Heredity 84: 127-143. [Abstract] [PDF]
- RS Mehta, AF Feder, SM Boca, NA Rosenberg (2019) The relationship between haplotype-based FST and haplotype length. Genetics 213: 281-295. [Abstract] [PDF] [Supplementary Figure 1] [Supplementary Figure 2] [Supplementary Figure 3] [Supplementary Figure 4]
- N Alcala, NA Rosenberg (2019) G'ST, Jost's D, and FST are similarly constrained by allele frequencies: a mathematical, simulation, and empirical study. Molecular Ecology 28: 1624-1636. [Abstract] [PDF] [Supplement]
- AJ Aw, NA Rosenberg (2018) Bounding measures of genetic similarity and diversity using majorization. Journal of Mathematical Biology 77: 711-737. [Abstract] [PDF]
- N Alcala, NA Rosenberg (2017) Mathematical constraints on FST: biallelic markers in arbitrarily many populations. Genetics 206: 1581-1600. [Abstract] [PDF] [File S1] [File S2]
- NR Garud, NA Rosenberg (2015) Enhancing the mathematical properties of new haplotype homozygosity statistics for the detection of selective sweeps. Theoretical Population Biology 102: 94-101. [Abstract] [PDF]
- MD Edge, NA Rosenberg (2014) Upper bounds on FST in terms of the frequency of the most frequent allele and total homozygosity: the case of a specified number of alleles. Theoretical Population Biology 97: 20-34. [Abstract] [PDF]
- M Jakobsson, MD Edge, NA Rosenberg (2013) The relationship between FST and the frequency of the most frequent allele. Genetics 193: 515-528. [Abstract] [PDF]
- SB Reddy, NA Rosenberg (2012) Refining the relationship between homozygosity and the frequency of the most frequent allele. Journal of Mathematical Biology 64: 87-108. [Abstract] [PDF]
- SM Boca, NA Rosenberg (2011) Mathematical properties of Fst between admixed populations and their parental source populations. Theoretical Population Biology 80: 208-216. [Abstract] [PDF]
- NA Rosenberg, M Jakobsson (2008) The relationship between homozygosity and the frequency of the most frequent allele. Genetics 179: 2027-2036. [Abstract] [PDF]
- JM VanLiere, NA Rosenberg (2008) Mathematical properties of the r2 measure of linkage disequilibrium. Theoretical Population Biology 74: 130-137. [Abstract] [PDF]
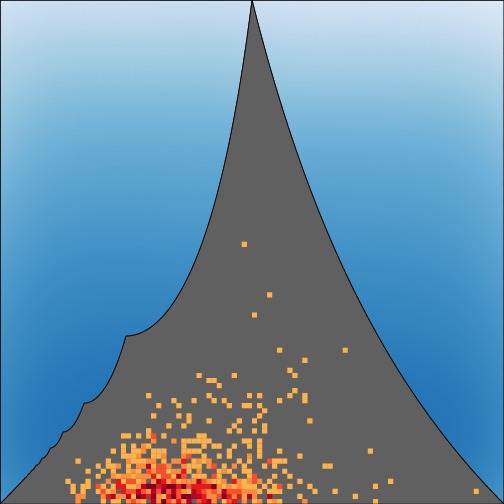
The strict upper bound
on the value of
FST at a locus given the frequency of the most
frequent
allele. See Jakobsson,
Edge, and Rosenberg (2013) for details.
Theoretical population genetics of admixture. When mating occurs between members of two or more groups that have long been separated, new populations can form that are admixed. Admixture is widespread in human populations, as a result of complex histories of migration, conquest, enslavement, and ongoing cultural interactions. A popular population-genetic model treats allele frequencies in an admixed population as linear combinations of the allele frequencies in its source populations, weighting each frequency by an admixture coefficient for its corresponding source population. We have examined a number of features of this admixture model in relation to the Fst measure of genetic differentiation, statistics for measuring ancestry information content, and neighbor-joining inference of population trees. Further, we have extended beyond the statistical model of admixture to develop a mechanistic model that acocunts for varying contributions of different source populations over time. This model enables assessments of the impact of different admixture histories on the pattern of admixture across individuals, and we are using it for analysis of the history and structure of admixture in a variety of admixed human populations.
- L Agranat-Tamir, KD Agwamba, JA Mooney, NA Rosenberg (2026) Shared ancestors and the birthday problem. American Statistician 80: 413-422. [Abstract] [PDF]
- L Agranat-Tamir, JA Mooney, NA Rosenberg (2024) Counting the genetic ancestors from source populations in members of an admixed population. Genetics 226: iyae011. [Abstract] [PDF] [Supplement]
- JA Mooney, L Agranat-Tamir, JK Pritchard, NA Rosenberg (2023) On the number of genealogical ancestors tracing to the source groups of an admixed population. Genetics 224: iyad079. [Abstract] [PDF] [Supplement]
- J Kim, MD Edge, A Goldberg, NA Rosenberg (2021) Skin deep: the decoupling of genetic admixture levels from phenotypes that differed between source populations. American Journal of Physical Anthropology 175: 406-421. [Abstract]
- SM Boca, L Huang, NA Rosenberg (2020) On the heterozygosity of an admixed population. Journal of Mathematical Biology 81: 1217-1250. [Abstract]
- A Goldberg, A Rastogi, NA Rosenberg (2020) Assortative mating by population of origin in a mechanistic model of admixture. Theoretical Population Biology 134: 129-146. [Abstract]
- J Kim, F Disanto, NM Kopelman, NA Rosenberg (2019) Mathematical and simulation-based analysis of the behavior of admixed taxa in the neighbor-joining algorithm. Bulletin of Mathematical Biology 81: 452-493. [Abstract] [PDF] [Supplement]
- NA Rosenberg (2016) Admixture models and the breeding systems of H. S. Jennings: a GENETICS connection. Genetics 202: 9-13. [PDF]
- A Goldberg, NA Rosenberg (2015) Beyond 2/3 and 1/3: the complex signatures of sex-biased admixture on the X chromosome. Genetics 201: 263-279. [Abstract] [PDF]
- EO Buzbas, NA Rosenberg (2015) AABC: approximate approximate Bayesian computation for inference in population-genetic models. Theoretical Population Biology 99: 31-42. [Abstract] [PDF]
- A Goldberg, P Verdu, NA Rosenberg (2014) Autosomal admixture levels are informative about sex bias in admixed populations. Genetics 198: 1209-1229. [Abstract] [PDF]
- NM Kopelman, L Stone, O Gascuel, NA Rosenberg (2013) The behavior of admixed populations in neighbor-joining inference of population trees. Pacific Symposium on Biocomputing 18: 273-284. [Abstract] [PDF]
- P Verdu, NA Rosenberg (2011) A general mechanistic model for admixture histories of hybrid populations. Genetics 189: 1413-1426. [Abstract] [PDF] [Supplementary Text] [Supplementary Figure 1] [Supplementary Table 1]
- SM Boca, NA Rosenberg (2011) Mathematical properties of Fst between admixed populations and their parental source populations. Theoretical Population Biology 80: 208-216. [Abstract] [PDF]
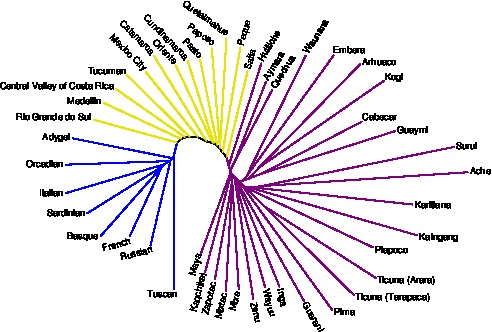
A neighbor-joining tree illustrating the interior placement of admixed populations in relation to populations from source regions. See Kopelman, Stone, Gascuel, and Rosenberg (2013) for details.
Human migration and spatial expansion. The genomes of living humans carry information about past human migrations. Patterns of genetic diversity and similarity among individuals and populations reflect a complex history of such phenomena as migration, natural selection, and changes in population size. As population-genetic models of migration and spatial expansion make predictions about extant genetic variation given assumptions about active evolutionary phenomena, they can help to understand the connection between extant genetic variation and past evolutionary processes. We have been developing and studying models of population migration with the aim of understanding the processes that have been active during human evolution. Examples include assessments of global models of human migration, evaluations of spatial patterns of genetic variation, and approaches for making use of genome-scale data.
- G Greenbaum, WM Getz, NA Rosenberg, MW Feldman, E Hovers, O Kolodny (2019) Disease transmission and introgression can explain the long-lasting contact zone of modern humans and Neanderthals. Nature Communications 10: 5003. [Abstract] [PDF] [Supplement]
- M DeGiorgio, NA Rosenberg (2013) Geographic sampling scheme as a determinant of the major axis of genetic variation in principal components analysis. Molecular Biology and Evolution 30: 480-488. [Abstract] [PDF]
- TJ Pemberton, D Absher, MW Feldman, RM Myers, NA Rosenberg, JZ Li (2012) Genomic patterns of homozygosity in worldwide human populations. American Journal of Human Genetics 91: 275-292. [Abstract] [PDF] [Main Supplement] [Supplementary Table 2 (.zip)] [Supplementary Table 3 (.zip)] [Supplementary Table 4 (.zip)] [Supplementary Table 5 (.zip)]
- NA Rosenberg (2011) A population-genetic perspective on the similarities and differences among worldwide human populations. Human Biology 83: 659-684. [Abstract] [PDF]
- M DeGiorgio, JH Degnan, NA Rosenberg (2011) Coalescence-time distributions in a serial founder model of human evolutionary history. Genetics 189: 579-593. [Abstract] [PDF]
- ZA Szpiech, NA Rosenberg (2011) On the size distribution of private microsatellite alleles. Theoretical Population Biology 80: 100-113. [Abstract] [PDF]
- C Wang, ZA Szpiech, JH Degnan, M Jakobsson, TJ Pemberton, JA Hardy, AB Singleton, NA Rosenberg (2010) Comparing spatial maps of human population-genetic variation using Procrustes analysis. Statistical Applications in Genetics and Molecular Biology 9: 13. [Abstract] [PDF]
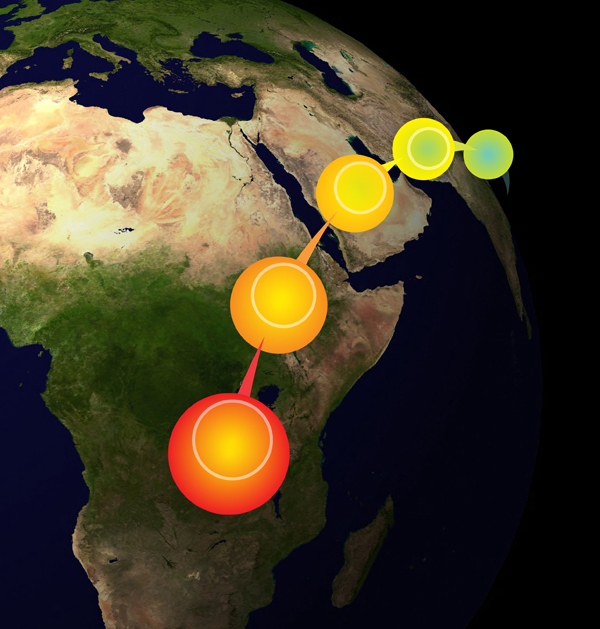
A schematic of a serial founder model for human migrations out of Africa. See the work of Degiorgio, Degnan, and Rosenberg (2011) for details.
Consanguinity, identity by descent, relatedness, and runs of homozygosity. Genomic data enable new approaches for studies of genetic relationships and patterns of individual genomic sharing. For example, human individuals possess long stretches of their genomes in which the genomic copies inherited from their two parents are genetically identical. These runs of homozygosity (ROH) reflect a variety of different processes, such as pairing of identical ancient haplotypes, background levels of relatedness among individuals within in a population, and recent parental relatedness. We have been characterizing runs of homozygosity, their differences across human populations, and their connection with such processes as inbreeding, linkage disequilibrium, and the amplification of deleterious variation. We are also devising new approaches for assessing patterns of variation in data sets with a high level of genetic relatedness. Studies of ROH and relatedness contribute to such topics as clinical genomic testing, conservation genetics, and identification of genes for rare recessive diseases.
- E Heinrich Mora, NA Rosenberg (2026) An nth-cousin mating model and the n-anacci numbers. Fibonacci Quarterly 64: 270-280. [Abstract] [PDF]
- DJ Cotter, AL Severson, JTL Kang, HN Godrej, S Carmi, NA Rosenberg (2024) Modeling the effects of consanguinity on autosomal and X-chromosomal runs of homozygosity and identity-by-descent sharing. G3: Genes, Genomes, Genetics 14: jkad264. [Abstract] [PDF] [Supplement]
- DJ Cotter, AL Severson, S Carmi, NA Rosenberg (2022) Limiting distribution of X-chromosomal coalescence times under first-cousin consanguineous mating. Theoretical Population Biology 147: 1-15. [Abstract]
DJ Cotter, AL Severson, NA Rosenberg The effect of consanguinity on coalescence times on the X chromosome. Theoretical Population Biology 140: 32-43 (2021). [Abstract]
- AL Severson, S Carmi, NA Rosenberg (2021) Variance
and limiting distribution of coalescence times in a diploid model of a
consanguineous population. Theoretical Population Biology
139: 50-65. [Abstract]
- AL Severson, S Carmi, NA Rosenberg (2019) The effect of consanguinity on between-individual identity-by-descent sharing. Genetics 212: 305-316. [Abstract] [PDF]
- J Kim, MD Edge, BFB Algee-Hewitt, JZ Li, NA Rosenberg (2018) Statistical detection of relatives typed with disjoint forensic and biomedical loci. Cell 175: 848-858. [Abstract] [PDF] [Supplement]
- NA Rosenberg, TJ Pemberton, JZ Li, JW Belmont (2013) Runs of homozygosity and parental relatedness. Genetics in Medicine 15: 753-754. [PDF]
- ZA Szpiech, J Xu, TJ Pemberton, W Peng, S Zöllner, NA Rosenberg, JZ Li (2013) Long runs of homozygosity are enriched for deleterious variation. American Journal of Human Genetics 93: 90-102. [Abstract] [PDF] [Supplement] [Data]
- TJ Pemberton, F-Y Li, EK Hanson, NU Mehta, S Choi, J Ballantyne, JW Belmont, NA Rosenberg, C Tyler-Smith, PI Patel (2012) Impact of restricted marital practices on genetic variation in an endogamous Gujarati group. American Journal of Physical Anthropology 149: 92-103. [Abstract] [PDF] [Supplement (.docx)] [Data]
- TJ Pemberton, D Absher, MW Feldman, RM Myers, NA Rosenberg, JZ Li (2012) Genomic patterns of homozygosity in worldwide human populations. American Journal of Human Genetics 91: 275-292. [Abstract] [PDF] [Main Supplement] [Supplementary Table 2 (.zip)] [Supplementary Table 3 (.zip)] [Supplementary Table 4 (.zip)] [Supplementary Table 5 (.zip)]
- M DeGiorgio*, I Jankovic*, NA Rosenberg (2010) Unbiased estimation of gene diversity in samples containing related individuals: exact variance and arbitrary ploidy. Genetics 186: 1367-1387. [Abstract] [PDF]
- TJ Pemberton, C Wang, JZ Li, NA Rosenberg (2010) Inference of unexpected genetic relatedness among individuals in HapMap Phase III. American Journal of Human Genetics 87: 457-464. [Abstract] [PDF] [Supplement]

The 203 configurations of identity and nonidentity possible among the six alleles at a locus in three diploid individuals. The figure is inspired by the work of Degiorgio, Jankovic, and Rosenberg (2010).
Combinatorics of evolutionary trees. Evolution within populations gives rise to trees of genetic lineages. When multiple species related by a species tree are considered, gene trees can differ in topology from each other and from the species tree on which they evolve. The joint analysis of gene trees and species trees then gives rise to consideration of a number of characteristic mathematical objects, such as coalescent histories and deep coalescences. Given a gene tree and a species tree, a coalescent history is a list of the branches of the species tree on which coalescences in the gene tree take place. A deep coalescence is tabulated when a pair of gene lineages fail to coalesce along a branch of the species tree. We have been examining how coalescent histories, deep coalescences, and other combinatorial features of gene trees and species trees generate both problems of mathematical interest as well as insights into the development and performance of methods for the inference of species trees.
L Agranat-Tamir, M Fuchs, B Gittenberger, NA Rosenberg, KV Seetharaman (2026) Combinatorial comparison of general galled trees, time-consistent galled trees, and simplex time-consistent galled trees. Advances in Applied Mathematics 180: 103131. [Abstract]
- L Agranat-Tamir, M Fuchs, B Gittenberger, NA Rosenberg (2026) Enumerative combinatorics of unlabeled and labeled time-consistent galled trees. Theoretical Computer Science 1082: 116075. [Abstract]
- EH Dickey, NA Rosenberg (2026) Labeled histories and maximally probable labeled topologies with multifurcation. Discrete Applied Mathematics 391: 192-203. [Abstract] [PDF]
- CE Shiff, NA Rosenberg (2026) Enumeration of rooted binary perfect phylogenies. Discrete Applied Mathematics 380: 538-561. [Abstract] [PDF]
- L Devroye, MR Doboli, NA Rosenberg, S Wagner (2025) Tree height and the asymptotic mean of the Colijn-Plazzotta rank of unlabeled binary rooted trees. Bulletin of Mathematical Biology 87: 172. [Abstract] [PDF]
- EH Dickey, NA Rosenberg (2025) Labeled histories with multifurcation and simultaneity. Philosophical Transactions of the Royal Society B Biological Sciences 380: 20230307. [Abstract] [PDF]
- L Agranat-Tamir, M Fuchs, B Gittenberger, NA Rosenberg (2024) Asymptotic enumeration of rooted binary unlabeled galled trees with a fixed number of galls. In C. Mailler, S. Wild, eds. Proceedings of the 35th International Conference on Probabilistic, Combinatorial and Asymptotic Methods for the Analysis of Algorithms (AofA 2024). Leibniz International Proceedings in Informatics (LIPIcs) 302: 27. Schloss Dagstuhl — Leibniz-Zentrum für Informatik. [Abstract] [PDF]
- M Doboli, H-K Hwang, NA Rosenberg (2024) Periodic behavior of the minimal Colijn-Plazzotta rank for trees with a fixed number of leaves. In C. Mailler, S. Wild, eds. Proceedings of the 35th International Conference on Probabilistic, Combinatorial and Asymptotic Methods for the Analysis of Algorithms (AofA 2024). Leibniz International Proceedings in Informatics (LIPIcs) 302: 18. Schloss Dagstuhl — Leibniz-Zentrum für Informatik. [Abstract] [PDF]
- L Agranat-Tamir, S Mathur, NA Rosenberg (2024) Enumeration of rooted binary unlabeled galled trees. Bulletin of Mathematical Biology 86: 45. [Abstract] [PDF]
- E Lappo, NA Rosenberg (2024) A lattice structure for ancestral configurations arising from the relationship between gene trees and species trees. Discrete Applied Mathematics 343: 65-81. [Abstract] [PDF]
- ARP Maranca, NA Rosenberg (2024) Bijections between the multifurcating unlabeled rooted trees and the positive integers. Advances in Applied Mathematics 153: 102612. [Abstract] [PDF].
- F Disanto, M Fuchs, C-Y Huang, AR Paningbatan, NA Rosenberg (2024) The distributions under two species-tree models of the total number of ancestral configurations for matching gene trees and species trees. Advances in Applied Mathematics 152: 102594. [Abstract]
- MC King, NA Rosenberg (2023) A mathematical connection between single-elimination sports tournaments and evolutionary trees. Mathematics Magazine 96: 484-497. [Abstract] [PDF]
- S Mathur, NA Rosenberg (2023) All galls are divided into three or more parts: recursive enumeration of labeled histories for galled trees. Algorithms for Molecular Biology 18:1. [Abstract] [PDF]
- F Disanto, M Fuchs, AR Paningbatan, NA Rosenberg (2022) The distributions under two species-tree models of the number of root ancestral configurations for matching gene trees and species trees. Annals of Applied Probability 32: 4426-4458. [Abstract] [PDF]
- JA Palacios, A Bhaskar, F Disanto, NA Rosenberg (2022) Enumeration of binary trees compatible with a perfect phylogeny. Journal of Mathematical Biology 84: 54. [Abstract] [PDF]
- MC King, NA Rosenberg (2021) A simple derivation of the mean of the Sackin index of tree balance under the uniform model on rooted binary labeled trees. Mathematical Biosciences 342: 108688. [PDF]
-
E Alimpiev, NA Rosenberg Enumeration of coalescent histories for caterpillar species trees and p-pseudocaterpillar gene trees. Advances in Applied Mathematics 131: 102265 (2021). [Abstract] [PDF]
- NA Rosenberg (2021) On the Colijn-Plazzotta numbering scheme for unlabeled binary rooted trees. Discrete Applied Mathematics 291: 88-98. [Abstract] [PDF]
- J Kim, NA Rosenberg, JA Palacios (2020) Distance metrics for ranked evolutionary trees. Proceedings of the National Academy of Sciences 117: 28876-28886. [Abstract] [PDF] [Supplement]
- ZM Himwich, NA Rosenberg (2020) Roadblocked monotonic paths and the enumeration of coalescent histories for non-matching caterpillar gene trees and species trees. Advances in Applied Mathematics 113: 101939. [Abstract]
- F Disanto, NA Rosenberg (2019) Enumeration of compact coalescent histories for matching gene trees and species trees. Journal of Mathematical Biology 78: 155-188. [Abstract] [PDF]
- F Disanto, NA Rosenberg (2019) On the number of non-equivalent ancestral configurations for matching gene trees and species trees. Bulletin of Mathematical Biology 81: 384-407. [Abstract] [PDF]
- NA Rosenberg (2019) Enumeration of lonely pairs of gene trees and species trees by means of antipodal cherries. Advances in Applied Mathematics 102: 1-17. [Abstract] [PDF]
- F Disanto, NA Rosenberg (2017) Enumeration of ancestral configurations for matching gene trees and species trees. Journal of Computational Biology 24: 831-850. [Abstract] [PDF]
- F Disanto, NA Rosenberg (2016) Asymptotic properties of the number of matching coalescent histories for caterpillar-like families of species trees. IEEE/ACM Transactions on Computational Biology and Bioinformatics 13: 913-925. [Abstract] [PDF]
- F Disanto, NA Rosenberg (2015) Coalescent histories for lodgepole species trees. Journal of Computational Biology 22: 918-929. [Abstract] [PDF]
- F Disanto, NA Rosenberg (2014) On the number of ranked species trees producing anomalous ranked gene trees. IEEE/ACM Transactions on Computational Biology and Bioinformatics 11: 1229-1238. [Abstract] [PDF]
- CV Than, NA Rosenberg (2014) Mean deep coalescence cost under exchangeable probability distributions. Discrete Applied Mathematics 174: 11-26. [Abstract] [PDF]
- NA Rosenberg (2013) Coalescent histories for caterpillar-like families. IEEE/ACM Transactions on Computational Biology and Bioinformatics 10: 1253-1262. [Abstract] [PDF]
- NA Rosenberg (2013) Discordance of species trees with their most likely gene trees: a unifying principle. Molecular Biology and Evolution 30: 2709-2713. [Abstract] [Full-text at journal website] [PDF]
- CV Than, NA Rosenberg (2013) Mathematical properties of the deep coalescence cost. IEEE/ACM Transactions on Computational Biology and Bioinformatics 10: 61-72. [Abstract] [PDF]
- JH Degnan, NA Rosenberg, T Stadler (2012) A characterization of the set of species trees that produce anomalous ranked gene trees. IEEE/ACM Transactions on Computational Biology and Bioinformatics 9: 1558-1568. [Abstract] [PDF]
- JH Degnan, NA Rosenberg, T Stadler (2012) The probability distribution of ranked gene trees on a species tree. Mathematical Biosciences 235: 45-55. [Abstract] [PDF]
- NA Rosenberg, JH Degnan (2010) Coalescent histories for discordant gene trees and species trees. Theoretical Population Biology 77: 145-151. [Abstract] [PDF]
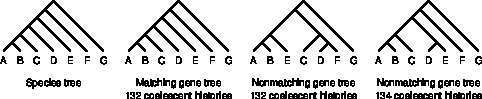
A gene tree that disagrees with the species tree can have as many or more coalescent histories than a matching gene tree. See Rosenberg & Degnan (2010) for details.
Coalescent theory. The coalescent, a stochastic process that connects genealogical lineages to a common ancestor through a process of "coalescence" of lineage pairs, represents a natural framework for studying the evolutionary history underlying a genetic sample. We have been developing coalescent-based models to investigate a variety of population-genetic phenomena, particularly in a setting in which multiple populations are themselves related through a common ancestral population. Areas of recent interest have been in the use of the coalescent in genotype imputation for genetic association studies, coalescent theory for the study of human evolution, and the coalescent along the branches of a phylogenetic tree.
- E Lappo, NA Rosenberg (2025) Coalescent theory of the ψ directionality index. G3: Genes, Genomes, Genetics 15: jkaf202. [Abstract] [PDF]
- DJ Cotter, AL Severson, JTL Kang, HN Godrej, S Carmi, NA Rosenberg (2024) Modeling the effects of consanguinity on autosomal and X-chromosomal runs of homozygosity and identity-by-descent sharing. G3: Genes, Genomes, Genetics 14: jkad264. [Abstract] [PDF] [Supplement]
- E Lappo, NA Rosenberg (2022) Approximations to the expectations and variances of ratios of tree properties under the coalescent. G3: Genes, Genomes, Genetics 12: jkac205. [Abstract] [PDF]
- DJ Cotter, AL Severson, S Carmi, NA Rosenberg (2022) Limiting distribution of X-chromosomal coalescence times under first-cousin consanguineous mating. Theoretical Population Biology 147: 1-15. [Abstract]
- RS Mehta, M Steel, NA Rosenberg (2022) The probability of joint monophyly of samples of gene lineages for all species in an arbitrary species tree. Journal of Computational Biology 27: 679-703. [Abstract]
- E Alimpiev, NA Rosenberg (2022) A compendium of covariances and correlation coefficients of coalescent tree properties. Theoretical Population Biology 143: 1-13. [Abstract] [PDF]
DJ Cotter, AL Severson, NA Rosenberg The effect of consanguinity on coalescence times on the X chromosome. Theoretical Population Biology 140: 32-43 (2021). [Abstract]
- AL Severson, S Carmi, NA Rosenberg (2021) Variance
and limiting distribution of coalescence times in a diploid model of a
consanguineous population. Theoretical Population Biology
139: 50-65. [Abstract]
- RS Mehta, NA Rosenberg (2019) The probability of reciprocal monophyly of gene lineages in three and four species. Theoretical Population Biology 129: 133-147. [Abstract] [PDF]
- N Alcala, A Goldberg, U Ramakrishnan, NA Rosenberg (2019) Coalescent theory of migration network motifs. Molecular Biology and Evolution 36: 2358-2374. [Abstract] [PDF] [Supplement]
- AL Severson, S Carmi, NA Rosenberg (2019) The effect of consanguinity on between-individual identity-by-descent sharing. Genetics 212: 305-316. [Abstract] [PDF]
- IM Arbisser, EM Jewett, NA Rosenberg (2018) On the joint distribution of tree height and tree length under the coalescent. Theoretical Population Biology 122: 46-56. [Abstract] [PDF]
- RS Mehta, D Bryant, NA Rosenberg (2016) The probability of monophyly of a sample of gene lineages on a species tree. Proceedings of the National Academy of Sciences USA 113: 8002-8009. [Abstract] [PDF] [Supplement] [Software]
- EM Jewett, NA Rosenberg (2014) Theory and applications of a deterministic approximation to the coalescent model. Theoretical Population Biology 93: 14-29. [Abstract] [PDF]
- L Huang, EO Buzbas, NA Rosenberg (2013) Genotype imputation in a coalescent model with infinitely-many-sites mutation. Theoretical Population Biology 87: 62-74. [Abstract] [PDF]
- EM Jewett*, M Zawistowski*, NA Rosenberg, S Zöllner (2012) A coalescent model for genotype imputation. Genetics 191: 1239-1255. [Abstract] [PDF]
- D Bryant, R Bouckaert, J Felsenstein, NA Rosenberg, A RoyChoudhury (2012) Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Molecular Biology and Evolution 29: 1917-1932. [Abstract] [PDF] [Supplement]
- JH Degnan, NA Rosenberg, T Stadler (2012) The probability distribution of ranked gene trees on a species tree. Mathematical Biosciences 235: 45-55. [Abstract] [PDF]
- M DeGiorgio, JH Degnan, NA Rosenberg (2011) Coalescence-time distributions in a serial founder model of human evolutionary history. Genetics 189: 579-593. [Abstract] [PDF]
- ZA Szpiech, NA Rosenberg (2011) On the size distribution of private microsatellite alleles. Theoretical Population Biology 80: 100-113. [Abstract] [PDF]
- NA Rosenberg, M Nordborg (2002) Genealogical trees, coalescent theory, and the analysis of genetic polymorphisms. Nature Reviews Genetics 3: 380-390. [Abstract] [PDF]
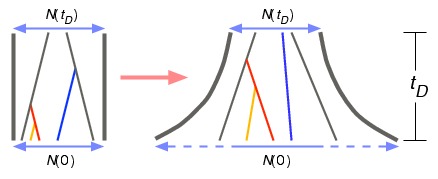
Population growth and coalescent waiting times. See Jewett, Zawistowski, Zöllner, and Rosenberg (2012) for details.
Inference of species trees under gene tree discordance. It has long been known that gene trees and species trees need not have the same shape. Surprisingly, we have found that gene tree discordance can be so great that under a standard model of within-species evolution, for any species tree topology with five or more species, there exist branch lengths for which the most likely gene tree topology to evolve along the branches of a species tree differs from the species phylogeny. This phenomenon of "anomalous gene trees" implies that when combining data on multiple loci, the simple procedure of using the most frequently observed gene tree topology to infer the species tree topology can be asymptotically guaranteed to produce an incorrect estimate. We have been exploring the properties of probability models for gene trees conditional on species trees, developing tools for inference of species trees in the setting of gene tree discordance, and analyzing their performance.
- A Kim, NA Rosenberg, JH Degnan (2020) Probabilities of unranked and ranked anomaly zones under birth-death models. Molecular Biology and Evolution 37: 1480-1494. [Abstract]
- LH Uricchio, T Warnow, NA Rosenberg (2016) An analytical upper bound on the number of loci required for all splits of a species tree to appear in a set of gene trees. BMC Bioinformatics 17: 417. [Abstract] [PDF]
- M DeGiorgio, NA Rosenberg (2016) Consistency and inconsistency of consensus methods for inferring species trees from gene trees in the presence of ancestral population structure. Theoretical Population Biology 110: 12-24. [Abstract] [PDF]
- D Bryant, R Bouckaert, J Felsenstein, NA Rosenberg, A RoyChoudhury (2012) Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Molecular Biology and Evolution 29: 1917-1932. [Abstract] [PDF] [Supplement]
- LJ Helmkamp, EM Jewett, NA Rosenberg (2012) Improvements to a class of distance matrix methods for inferring species trees from gene trees. Journal of Computational Biology 19: 632-649. [Abstract] [PDF]
- EM Jewett, NA Rosenberg (2012) iGLASS: an improvement to the GLASS method for estimating species trees from gene trees. Journal of Computational Biology 19: 293-315. [Abstract] [PDF]
- CV Than, NA Rosenberg (2011) Consistency properties of species tree inference by minimizing deep coalescences. Journal of Computational Biology 18: 1-15. [Abstract] [PDF]
- JH Degnan, M DeGiorgio, D Bryant, NA Rosenberg (2009) Properties of consensus methods for inferring species trees from gene trees. Systematic Biology 58: 35-54. [Abstract] [PDF]
- JH Degnan, NA Rosenberg (2009) Gene tree discordance,
phylogenetic inference and the multispecies coalescent. Trends in
Ecology and Evolution 24:
332-340. [Abstract]
[PDF]
[Supplement]
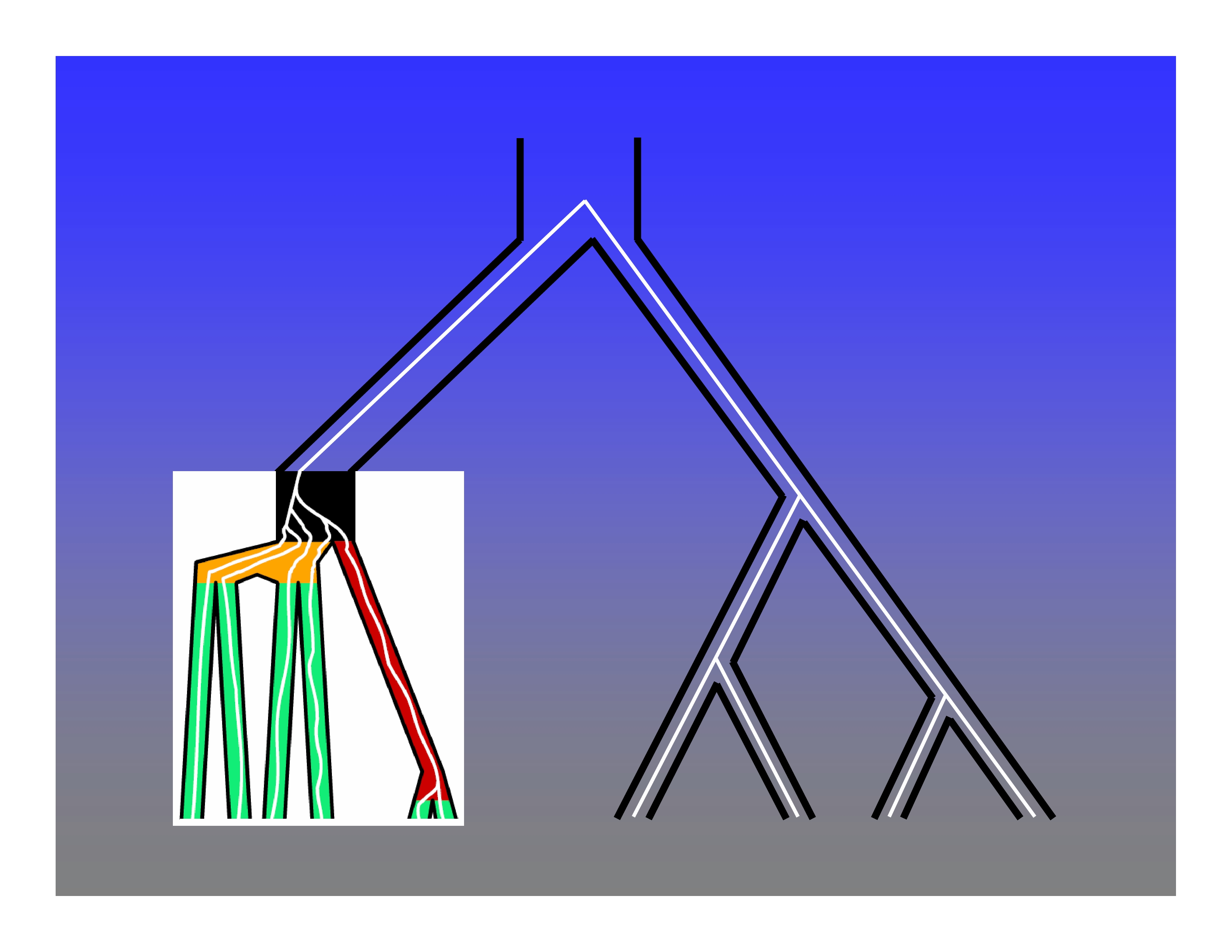
An inductive proof that all species tree topologies with five or more taxa have anomalous gene trees. See Degnan & Rosenberg (2006) and Rosenberg (2013) for details.